本篇介紹執行定序分析常見的報錯情境,
而報錯的方式千百種,查詢解決方法就一種 :錯誤訊息複製起來,扔到 Google 就對了
不過總有特別頑固又印象深刻的錯誤,
這邊分享幾種曾經遇過特別的報錯 :
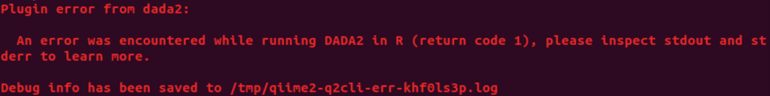
這應該是最常見也是頭最疼的一個報錯,
DADA2 是負責品質管制的 R 套件,
頭疼的原因是永遠報錯都是 return code 1(難道沒有其他數字了嗎 ! ),
不會直接顯示真正的錯誤訊息,
注意看會有一行紅字 :
Debug info has been saved to /tmp/XXXXXXXX.log
此時複製路徑並輸入 :
head /tmp/XXXXXXXX.log
就能夠一窺它壞掉的原因了Q,把裏頭真正錯誤訊息丟 Google 才找得到解法。
情境舉例 : 錯誤訊息 - Mismatched forward and reverse sequence files
'Filtering Error in (function (fn, fout, maxN = c(0, 0), truncQ = c(2, 2), truncLen = c(0, : Mismatched forward and reverse sequence files: 3296, 4292.'
這情況會發生在抓取公開文獻的序列資料來分析時遇到的 Bug ,
主因是 NGS 雙尾定序 (Pair-end)原始資料兩邊序列不等長,
為什麼會不等長可能要問問作者或神奇海螺,
解法如下 :
調出 QIIME2 錯誤報告資料夾
qiime tools export \
--input-path demux.qza \
--output-path debugging
切換到 debugging 資料夾
cd debugging
列出該批待處理樣本檔名與序列長度資訊
for f in *.fastq.gz; do r=$(( $(gunzip -c $f | wc -l | tr -d '[:space:]') / 4 )); echo $r $f; done
看起來會像是這樣 (示意圖) :
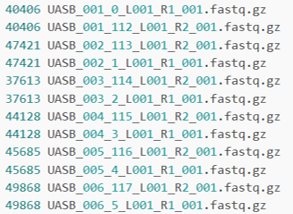
回去一開始觀察錯誤訊息,若有3296, 4292 出現在列表中,
建議直接到manifest.tsv 移除那對樣本,
不要分析它了,請學會放下它,執著並不符時間成本。
Reference : forum.qiime2.org
這是因為取樣深度 (sampling depth) 調得太高的關係,
觀念參閱 [Day 15] !
本篇文章同步刊載於科學毛怪部落格 PetSci Blog。


 iThome鐵人賽
iThome鐵人賽